Zhi Chen†, Eric Blanc†§ and Michael S. Chapman*†‡
* Department of Chemistry & † Institute of Molecular Biophysics, Florida State University, Tallahassee, FL 32306-4380, USA.
Synopsis: Torsion angle molecular dynamics makes real-space methods the most efficient for all but the final cycles of refinement when excellent (anomalous diffraction) phases are available. Overfitting is reduced and convergence improved by implicit use of phases and use of a local refinement that does not allow remote errors to be mutually compensating.
Abstract
Real-space targets and molecular dynamics search protocols have been combined to improve the convergence of macromolecular atomic refinement. This was accomplished by providing a local real-space target function for the molecular dynamics program X-plor (Brünger, A.T., 1992, X-Plor Version 3.1 A System for X-ray Crystallography and NMR, Yale University Press). With poor isomorphous replacement experimental phases, molecular dynamics does not improve real-space refinement. However, with high quality anomalous diffraction phases, convergence is improved at the start of refinement, and torsion angle real-space molecular dynamics performs better than other available least-squares or maximum likelihood methods in real- or reciprocal-space. It is shown that the improvements result from an optimization method that can escape local minima and from a reduction of overfitting through the implicit use of phases, and through use of a local refinement in which errors in remote parts of the structure can not be mutually compensating.
Introduction
The most common macromolecular crystallographic refinement involves restrained optimization of the agreement between diffraction amplitudes calculated from an atomic model and those that are derived from the experimental data (Jensen, 1985 ). Stereochemical restraints are introduced either by optimizing the agreement with ideal geometries (Engh & Huber, 1991; Hendrickson, 1985; Waser, 1963) or (as in this work) through minimization of an empirical estimate of the configurational potential energy, Echem. (Brünger et al., 1987; Levitt, 1974). Structural refinement then becomes minimization of the objective function:
| Equation 1 |
Although reciprocal-space Exray terms are currently preferred, early refinements used a real-space target (Diamond, 1971):
| Equation 2 |
| Equation 3 |
Like earlier implementations, the newer real-space refinements optimized the structure by gradient descent methods (Tronrud, 1992). This work examines the potential of combining two methods that have robust convergence properties – namely real-space targets and molecular dynamics optimization. The potential application of such methodology has broadened considerably recently with the availability of accurate macromolecular phases from multiwavelength anomalous diffraction (MAD) methods (Hendrickson & Ogata, 1997).
Methods
Real space MD was implemented by programming an alternative target function for X-plor (Brünger, 1992b) that provides the option of substituting Equation 2 for Exray in Equation 1. A prior implementation of a real-space target (Chapman, 1995) was adapted so that input and output was compatible with X-plor programs, data files and control scripts. The target value and its derivatives with respect to the atomic parameters are calculated with the new module and passed back to X-plor. Thus, all methods of optimization that have been applied in reciprocal-space (Brünger et al., 1997) can now be applied in real-space, including torsion angle or Cartesian MD (and conjugate gradient optimization).
Initial tests used simulated structure amplitudes, phases
and maps calculated from -amylase
inhibitor (Pflugrath et al., 1989) between 17 and 2 Å resolution.
Starting models were perturbed by varying amounts using molecular dynamics
at 600K followed by energy minimization, all in the absence of a Exray
term. The test refinement protocol involved 4 ps of torsion angle MD at
8000 K (now including an Exray term), 0.2 ps of quenching at
300 K in Cartesian space, then conjugate gradient energy minimization.
Slow-cooling protocols were tested but, as in reciprocal space torsion
angle refinements (Rice & Brünger, 1994), they proved to be inferior
to rapid quenching and are not considered further. With phases calculated
directly from the correct structure, a starting model with 1.43 Å
rms backbone error is refined to an error of 0.1 Å. The convergence
radius is 3.6 Å, as defined by the maximal backbone perturbation
that can still be refined to approximate the correct structure. With an
omit map (Bhat, 1988 ) calculated from the perturbed model, the radius
of convergence was 0.6 Å, indicating dependence of the method upon
the availability of experimental phases.
Real-space torsion angle molecular dynamics (RSTAMD) was tested in two systems with actual crystallographic data. HMG CoA reductase represented poor experimental phases and mannose binding protein A (MBPA) represented high quality experimental phases. HMG CoA reductase exemplified a large protein structure determination in which poor multiple isomorphous replacement (MIR) phases were improved by the application of non-crystallographic symmetry (NCS) in the actual structure determination (Lawrence et al., 1995). For the current tests, the NCS was ignored and the unrefined model of Lawrence et al. was refined against a barely interpretable MIR map.
MBPA exemplified high quality MAD phasing (Burling et al., 1996). The starting model was based on a 2.3 Šresolution homologous complex with a different lanthanide ion (Weis et al., 1991), with remodeling of seven disordered terminal residues into the 1.8 Šresolution MAD map (Burling et al., 1996), deletion of solvent molecules, resetting all B-factors to 15 Ų, stereochemical regularization, and rigid body reciprocal-space refinement against the 1.8 Šcryo-diffraction data (Burling et al., 1996). Additional tests started with the 1.8 Šresolution final refined structure (Burling et al., 1996).
The refinement methods that were compared using MBPA included the following. All methods were as implemented in X-plor (Brünger, 1992b). When molecular dynamics was used, the torsion angle implementation (Rice & Brünger, 1994) was used and was followed by conjugate gradient minimization of the objective function:
1b. Further locally refined the amino acids with the worst agreement with the electron density, as indicated by correlation coefficient (Jones et al., 1991; calculated according to Zhou et al., 1998).
| Equation 4 |
| Equation 5 |
| Equation 6 |
Results & Discussion
Application to the least favorable case of poor MIR phases in HMGCoA reductase showed that MD and conjugate gradient real-space performed near-identically. It was shown previously (Chapman & Blanc, 1997) that for poor phases, real-space refinement enhances but does not supersede reciprocal-space methods when the two are alternated in the initial cycles. Here, it is confirmed that real-space refinement is limited by the poor MIR phases and not the optimization algorithm. Thus, least-squares and MD are equally effective.
The potential with good phases is graphically illustrated in Figure 1. Real-space MD, unlike its gradient descent counterpart, is able to pass through an unfavorable configuration to find the best fit to the electron density. Quantitatively, it is clear from Table 1 that real-space molecular dynamics is the most powerful method for initial refinement when the map quality is good. Rfree drops about twice as much as with any of the currently available reciprocal-space methods, including maximum likelihood. (Smaller differences between maximum likelihood and least squares targets are seen here compared with those observed earlier (Pannu & Read, 1996) due to the high quality 2.3 Å MBPA starting model.) Coordinate error drops about 60% farther for RSTAMD than for the reciprocal-space methods. Real-space gradient descent is intermediate in performance between RSTAMD and reciprocal-space methods. The RSTAMD-refined model has an Rfree about twice as close to the target model as those produced by reciprocal-space refinements, indicating that some, but not all of the changes normally made by manual intervention have been accomplished automatically. Unlike HMGCoA reductase, the benefit of following initial real-space refinement with reciprocal-space refinement is at most marginal, presumably because the phase quality is not limiting at this stage of the MBPA refinement.
At the end of refinement, the indications are different (Table 2). Starting with the published, fully refined 1.8 Å MBPA structure (Burling et al., 1996), additional refinement in real-space using the MAD phases yields a structure with Rfree slightly higher than in reciprocal space (0.217 vs. 0.207). It is the MAD phases that are limiting, as is shown through substitution of phases calculated from the final model which allows real-space refinement to equal reciprocal-space (Rfree = 0.206, Table 2). Thus, there comes a point in refinement when the model errors become low enough that phase error limits real-space refinement, and reciprocal-space methods are indicated. With high quality MAD phases, this point is reached only for the final cycles.
While improvements of Rfree and backbone coordinate error during initial refinement are appreciable, substantial errors remain in side chain coordinates with all refinement methods (Table 1). Some improvement is made with a new algorithm that is possible with a local method of real-space refinement. The worst amino acids are identified according to correlation between model and experimental electron density (Zhou et al., 1998) and, as individual amino acids, are given additional cycles of RSTAMD. Applied to the 13 worst amino acids, the overall rms coordinate error is reduced from 0.64 to 0.52 Å (Table 1), but there is little change to the R-factors, because it is a small fraction of the weakest-scattering part of the molecule that is improved. The remaining error is mostly because of the selection of incorrect rotamers, due to the lack of ordered water molecules in the model, and due to the remaining need to make some corrections interactively rather than through the automatic refinement methods used. The local procedure helps at the initial stages of refinement when all B-factors are uniform, and the local method enables more appropriate scaling (and refinement) between the more disordered parts of the model and their weak electron density. Such a procedure could also be used to try to fix automatically some of the most egregious errors of a model as highlighted by other (stereochemical) indicators, although there will still be a need for visual inspection to correct, for example, register errors.
Table 1 gives some indications of how real-space refinement helps in all but the final cycles. Although the greatest drop in Rfree is seen with real-space refinement, the drop in crystallographic Rwork is less than or equal to the drop by the various reciprocal-space refinements, and substantially less than the commonly used amplitude-based MD refinement. Thus, in real-space, overfitting is substantially reduced. Overfitting, early in refinement, can also be reduced by appropriately accounting for model and data errors in the maximum likelihood formulation (Pannu & Read, 1996). The source of the improvement with real-space methods is fundamentally different. It is the improvement of data:parameter ratio through the use of implicit phase information, and also the use of a local refinement method (see later). In the case of MBPA, with low model error and high phase accuracy, the reduction of overfitting is greater with real-space methods than with maximum likelihood methods. The pseudo-real-space methods (numbers 6, 6b & 7) also incorporate phase information, are the best of the reciprocal-space methods, and reduce overfitting somewhat, but not as much as the true real-space algorithm. This may seem counter-intuitive, due to at least a superficial correspondence of the real- and reciprocal-space operations (Diamond, 1971; Silva & Rossmann, 1985). However, there are differences in the weighting and in the local / global nature of refinement. The closest correspondence is between Exray(r ) and Exray(A, B) (methods 1 & 6b), when both incorporate figure of merit weighting. The remaining difference between these targets is presumably due to the local nature of real-space refinement versus the global nature of all reciprocal space and pseudo-real-space methods. In global methods, all parts of the model move to decrease the discrepancy between Fo and Fc. Atoms may be moved away from their correct locations to reduce discrepancies due to remote errors in the model (Hodel et al., 1992), an incomplete description of solvent or missing macromolecular atoms. With a local real-space method, these components to the overfitting are eliminated, leading to improved refinement at the early stages.
Conclusion
Real-space refinement can be significantly enhanced with the addition of molecular dynamics optimization methods. With high quality phases and maps, the improvement over other refinement methods is substantial, until phase errors dominate over model errors in the final cycles. With low quality maps and phases, the method is benign. This suggests a pragmatic approach when the experimental phases are of uncertain quality – do all possible refinement in real-space, then complete refinement in reciprocal space. With the high quality phases and maps that are increasingly available with anomalous diffraction, the results reported here suggest that real-space refinement will become an increasingly important part of the efficient structure determination of a significant proportion of protein structures.
Acknowledgements
We thank Temple Burling, Axel Brünger, Martin Lawrence and Cynthia Stauffacher for access to the MBPA and HMGCoA reductase coordinates and diffraction data. This work was supported by the National Science Foundation (BIR94-18741). Modifications to Xplor that enable real-space molecular dynamics refinement will be available directly from the authors and/or in an upcoming version of the Molecular Simlations Inc. implementation of Xplor.
References
Adams, P. D., Pannu, N. S.,
Read, R. J. & Brünger, A. T. (1997). Cross-validated maximum-liklihood
enhances simulated annealing crystallographic refinement. Proceedings
of the National Academy of Sciences, USA 94, 5018-23.
Arnold, E. & Rossmann, M. G. (1988). The Use of Molecular-Replacement
Phases for the Refinement of the Human Rhinovirus 14 Structure. Acta
Crystallographica A44, 270-282.
Bhat, T. N. (1988 ). Calculation of an OMIT map. . J.
Appl. Cryst. 21 , 279-281 .
Blanc, E. & Chapman, M. S. (1997). RSRef:
Interactive real-space refinement with stereochemical restraints for use
during model-building. Journal of Applied Crystallography 30,
566-7.
Brünger, A. T. (1992a). Free R value: a novel statistical
quantity for assessing the accuracy of crystal structures. Nature
355, 472-5.
Brünger, A. T. (1992b). X-Plor Version 3.1 A
System for X-ray Crystallography and NMR, Yale University Press, New
Haven.
Brünger, A. T., Adams, P. D. & Rice, L. M. (1997).
New Applications of Simulated Annealing in Crystallographic Refinement.
in NATO Advanced Study Institute on Direct Methods for Solving Macromolecular
Structures, Erice, Italy , pp. 149-163. Kluwer, Dortrecht, Netherlands,
Erice, Italy
Brünger, A. T., Kuriyan, J. & Karplus, M. (1987).
Crystallographic R factor Refinement by Molecular Dynamics. Science
235, 458-60.
Brünger, A. T. & Rice, L. M. (1997). Crystallographic
Refinement by Simulated Annealing: Methods and Applications. Methods
in Enzymology 277, 243-69.
Burling, F. T., Weis, W. I., Flaherty, K. M. & Brünger,
A. (1996). Direct Observation of Protein Solvation and Discrete Disorder
Using Experimental Crystallographic Phases. Science 271,
72-77.
Chapman, M. S. (1995). Restrained Real-Space Macromolecular
Atomic Refinement using a New Resolution-Dependent Electron Density Function.
Acta Crystallographica A51, 69-80.
Chapman, M. S. & Blanc, E. (1997). Potential use
of Real Space Refinement in Protein Structure Determination. Acta Crystallographica
D53, 203-6.
Chapman, M. S. & Rossmann, M. G. (1996). Structural
Refinement of the DNA-containing Capsid of Canine Parvovirus using RSRef,
a Resolution-Dependent Stereochemically Restrained Real-Space Refinement
Method. Acta Crystallographica D52, 129-42.
Chen, Z., Blanc, E. & Chapman, M. S. (1998). Improved
free R-factors for the cross-validation of structures. Acta Crystallographica
accepted for publication.
Diamond, R. (1971). A Real-Space Refinement Procedure
for Proteins. Acta Crystallographica A27, 436-452.
Engh, R. A. & Huber, R. (1991). Accurate Bond and
Angle Parameters for X-ray Protein Structure Refinement. Acta Crystallographica
A47, 392-400.
Hendrickson, W. A. & Ogata, C. M. (1997). Phase Determination
from Multiwavelength Anomalous Diffraction Measurements. Methods in
Enzymology 276, 494-523.
Hendrickson, W. W. (1985). Stereochemically Restrained
Refinement of Macromolecular Structures. Methods in Enzymology 115,
252-270.
Hodel, A., Kim, S.-H. & Brünger, A. T. (1992).
Model Bias in Macromolecular Crystallography. Acta Cryst. A48,
851-58.
Jensen, L. H. (1985 ). Overview of Refinement in Macromolecular
Structure Analysis . Methods in Enzymology 115 , 227-234
.
Jones, T. A. & Kjeldgaard, M. (1997). Electron-Density
Map Interpretation. Methods in Enzymology 277, 173-208.
Jones, T. A. & Liljas, L. (1984). Crystallographic
Refinement of Macromolecules having Non-crystallographic Symmetry. Acta
Crystallographica A 40, 50-7.
Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard,
M. (1991). Improved Methods for Building Protein Models in Electron Density
Maps and the Location of Errors in these Models. Acta Crystallographica
A47, 110-9.
Kleywegt, G. J. & Jones, T. A. (1997). Model Building
and Refinement Practice. Methods in Enzymology 277, 208-230.
Lawrence, C. M., Rodwell, V. M. & Stauffacher, C.
V. (1995). The crystal structure of Pseudomonoas mevalonii HMG-CoA
reductase at 3.0 Å resolution. Science 268, 1758-62.
Levitt, M. (1974). Energy Refinement of Hen Egg-White
Lysozyme. Journal of Molecular Biology 82, 393-420.
Murshudov, G., Vagin, A. & Dodson, E. (1997). Refinement
of Macromolecular Structures by the Maximum-Likelihood Method. Acta
Crystallographica D53, 240-255.
Pannu, N. S. & Read, R. J. (1996). Improved Structure
Refinement Through Maximum Likelihood. Acta Crystallographica A52,
659-668.
Pannu, N. S. & Read, R. J. (1997). Incorporation
of prior phase information improves maximum liklihood structural refinement.
in NATO Advanced Study Institute on Direct Methods for Solving Macromolecular
Structures, Erice, Italy , pp. 488. Kluwer, Dortrecht, Netherlands,
Erice, Italy
Pflugrath, J. W., Wiegand, G., Huber, R. & Vertesey,
L. (1989). Crystal structure determination, refinement, and the molecular
model of the a-amylase inhibitor Hoe-467A. Journal
of Molecular Biology 189, 383-6.
Rees, D. C. & Lewis, M. (1983). Incorporation of
Experimental Phases in a Restrained Refinement. Acta Crystallographica
A39, 94-97.
Rice, L. M. & Brünger, A. T. (1994). Torsion
Angle Dynamics: Reduced Variable Conformational Sampling Enhances Crystallographic
Structure Refinment. Proteins: Structure, Function and Genetics
19, 277-90.
Silva, A. M. & Rossmann, M. G. (1985). The Refinement
of Southern Bean Mosaic Virus in Reciprocal Space. Acta Crystallographica
B41, 147-57.
Tronrud, D. E. (1992). Conjugate-Direction Minimization:
an Improved Method for the Refinement of Macromolecules. Acta Crystallographica
A48, 912-916.
Waser, J. (1963). Least-Squares Refinement with Subsidiary
Conditions. Acta Crystallographica 16, 1091-4.
Weis, W. I., Kahn, R., Fourne, R., Drickamer, K. &
Hendrickson, W. A. (1991). Structure of the Calcium-Dependent Lectin Domain
from a Rat Mannose-Binding Protein Determined by MAD Phasing. Science
254, 1608-15.
Zhou, G., Wang, J., Blanc, E. & Chapman, M. S. (1998).
Determination of the Relative Precision of Atoms in a Macromolecular Structure.
Acta Crystallographica D54, 391-9.
Captions
Figure 1: Rotamer correction by real-space molecular dynamics refinement. Ile 147 of Mannose Binding Protein A (Burling et al., 1996) was perturbed to an incorrect rotamer using the modeling program O (Kleywegt & Jones, 1997). Refinement against the MAD experimental electron density (shown) corrects the rotamer. While this particular type of error might be correctable with the tools in O (Kleywegt & Jones, 1997), quick application of the refinement to the whole protein can substantially reduce the number of corrections that need to be made with an interactive modeling program.
Figures
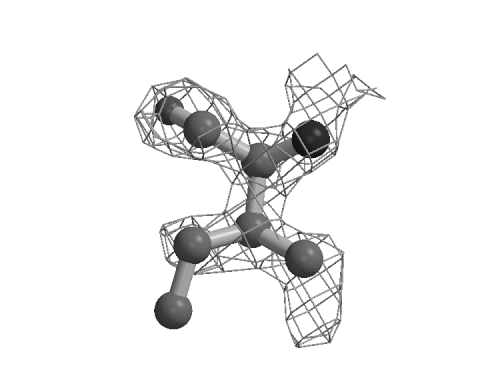
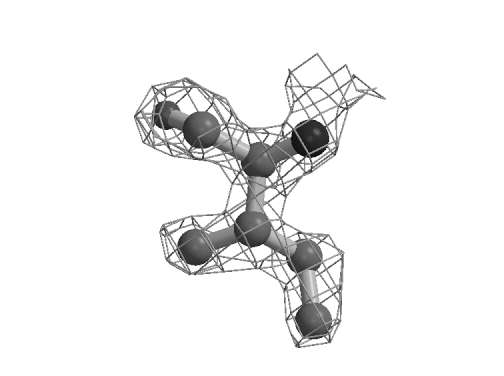
Figure 1
Tables
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
||||||
| Target: published 1.8 Å structure, less solvent & with B = 15. |
|
|
|
|
|||||
| Starting model (modified 2.3 Å structure). |
|
|
|
|
|||||
| 1 | Exray(r ) | Equation 2 |
|
|
|
|
|
||
| 1b | Exray(r ) | With local improvement* |
|
|
|
|
|
||
| 2 | Exray(r ) | Equation 2 |
|
|
|
|
|
||
| 3 | Exray(F) | Equation 1 |
|
|
|
|
|
||
| 4 | Exray(F) | Equation 1 |
|
|
|
|
|
||
| 5 | Exray(r ) then Exray(F) | Equation 2, Equation 1 |
|
|
|
|
|
||
| 6 | Exray(A,B) | Equation 4 |
|
|
|
|
|
||
| 6b | Exray(A,B) | With fom-weighting |
|
|
|
|
|
||
| 7 | Exray(F,f) | Equation 5 |
|
|
|
|
|
||
| 8 | Equation 6 |
|
|
|
|
|
|||
| 9 | Equation 6 |
|
|
|
|
|
|||
| 10 | + phase restraints |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
| Starting model (published 1.(Burling et al., 1996)[Burling, 1996 #991]). |
|
|
|
|||||
| 1 | Exray(r ) | Equation 2 |
|
|
|
|
|
|
| 3 | Exray(F) | Equation 1 |
|
|
|
|
||
| 1 | Exray(r ) | Equation 2 |
|
|
|
|
||